Understanding the Variant Listing¶
Advanced report findings are displayed in the Variant listing, which provides information about each variant to help determine whether the variant might be causative of the condition being analyzed in the selected study. Findings are organized in a series of tabs, and can be sorted, filtered, and annotated further using a selection of tools and options.
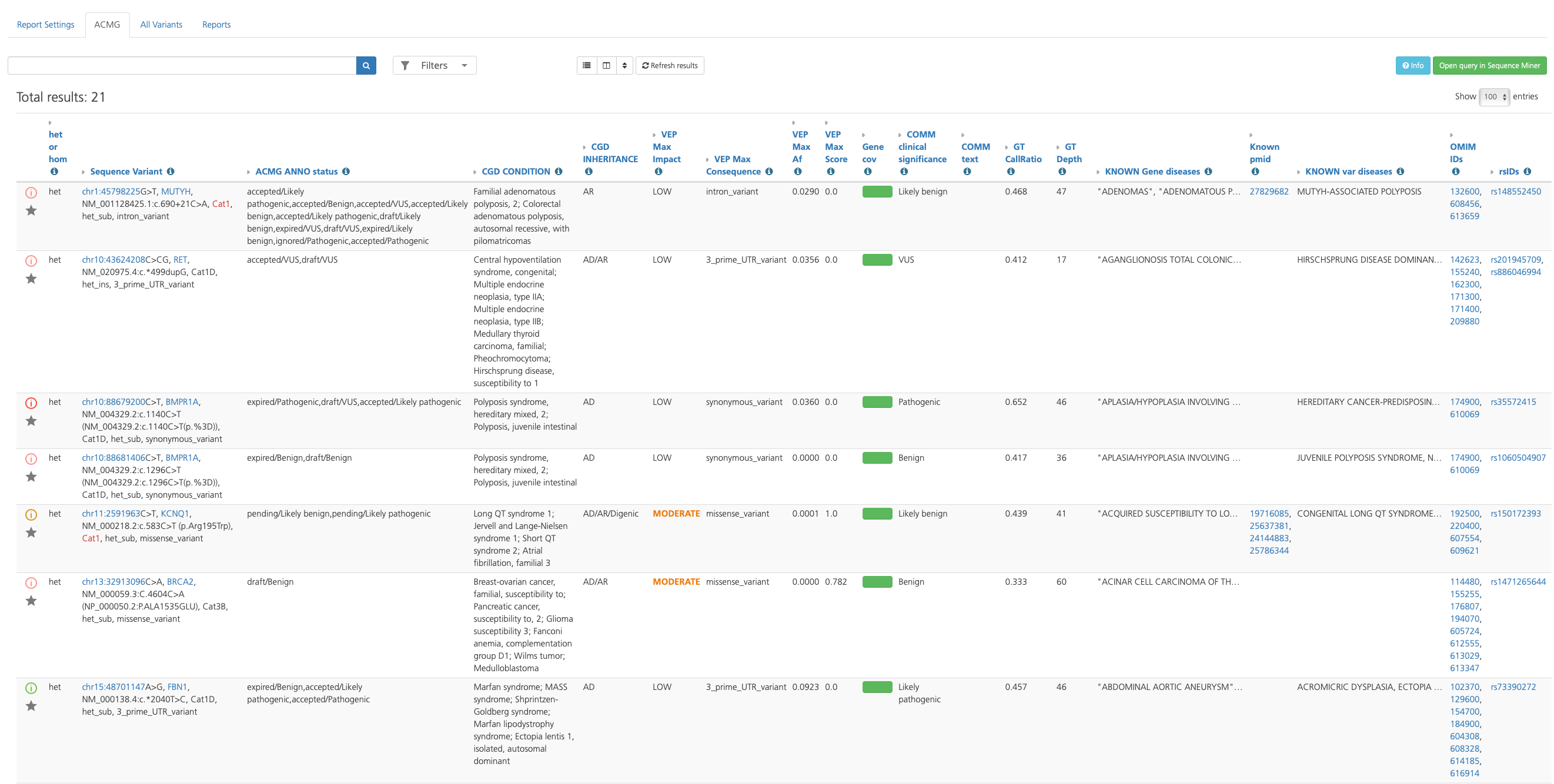
Variant listing in the Advanced report¶
Toggling views¶
The Variant listing opens in Table view by default, but findings on select tabs (e.g., Variants and Missing) can be viewed in either Table view or List view by toggling the Table/List icon at the top of the page. Both views display similar attributes, but Table view columns can be further customized.
To toggle between views, click the Table/List view icon at the top of the page.
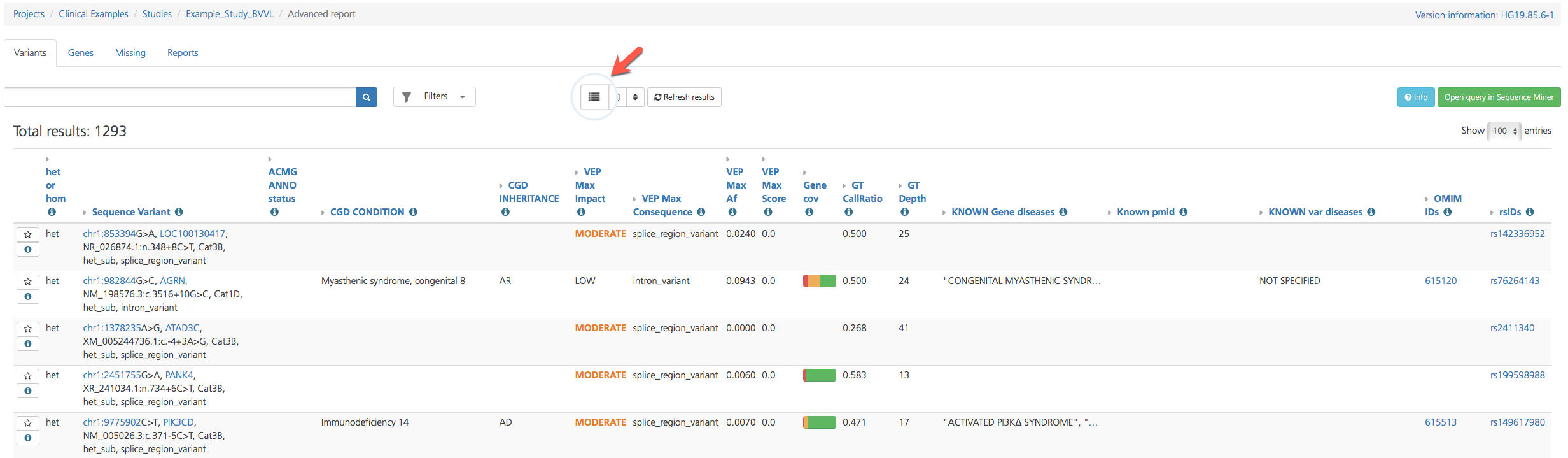
Variant listing in Table view¶
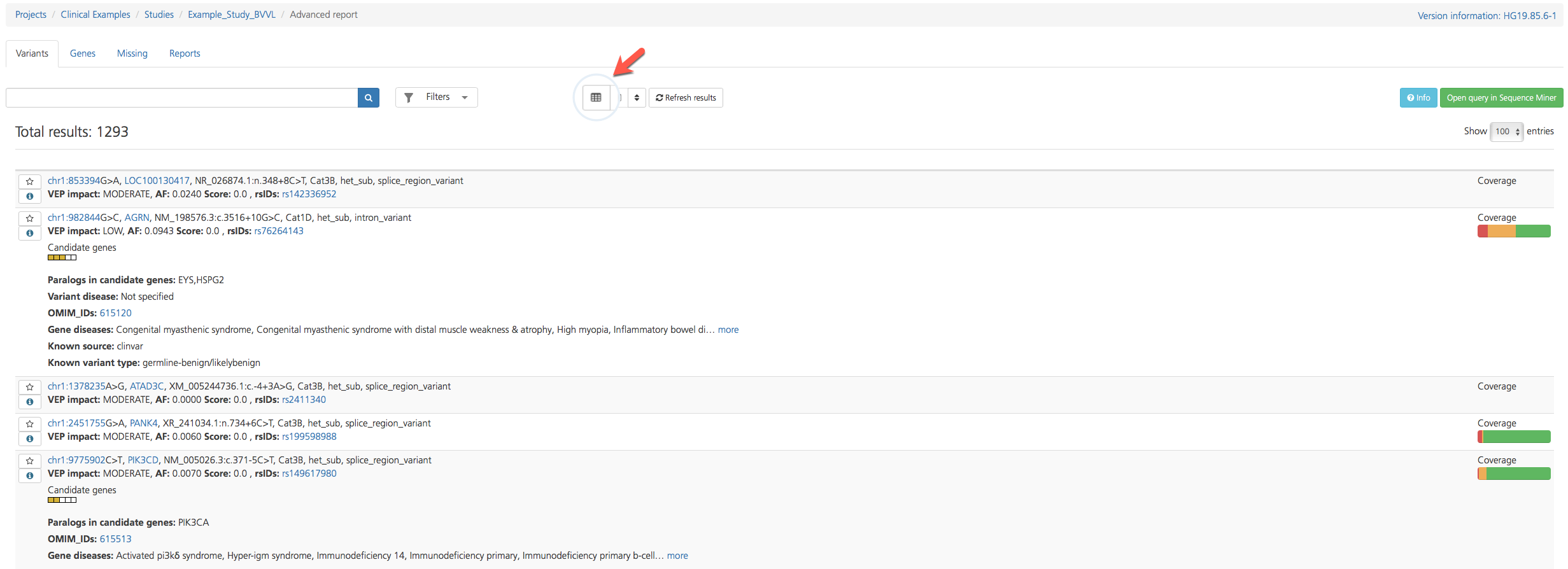
Variant listing in List view¶
Table view¶
Because there are too many annotations to fit into a single view, the Default columns displayed in Table view contain only a subset of the total available columns. Columns are broadly categorized by variant attribute categories and can be customized by clicking the Choose columns icon at the top of the page to show or hide selected columns (see also Filtering Results).
Default columns provide basic sequence variant call information, as well as clinically relevant annotations, links to external reference sources, and quality measures. Default columns are described in the table below. For a full list of column descriptions, see Advanced Report Columns.
Column |
Description |
|---|---|
Default |
|
het or hom |
Heterozygous or homozygous variant at the designated position |
Sequence Variant |
Condensed summary description of the variant combining data from other columns including variant location, gene, reference variant, called variant, coding sequence and protein position, and associated disease information (see also Sequence Variant) |
CGD CONDITION |
Conditions resulting from variations in the same gene that may otherwise be placed in the “General” intervention category |
CGD INHERITANCE |
Patterns of inheritance, including:
|
VEP Max Impact |
The classification of the variant based on the severity of the consequence type on the transcript (“HIGH”, “MODERATE”, “LOW”, “LOWEST”); for a given variant, the maximum observed impact across the predicted transcripts from this position is reported (see also Variant Effect Predictor (VEP) Settings) |
VEP Max Consequence |
The consequence type reported for this variant that has the greatest biological impact; for a given variant, the maximum observed consequence across the predicted transcripts from this position is reported (see also Variant Effect Predictor (VEP) Settings) |
VEP Max Af |
Maximum reported population allele frequency derived from the 1000 Genomes Project phase 3 (1000G3), Exome Variant Server (EVS), Exome Aggregation Consortium (ExAC), Genomes of the Netherlands (GONL), and Kyoto population surveys (see also popAlleleFreq) |
VEP Max Score |
Maximum score for the variant as observed in dbNSFP (Score=max of (1-Sift_score), Polyphen2_HDIV_score, or Polyphen2_HVAR_score) |
Gene cov |
Fraction of genes (exome) with depths <5 (red), ranging from 5-9 (orange), and greater than 9 (green); note that the scale ranges from 0 to 50% to make the red/orange bars longer, emphasizing low coverages |
GT CallRatio |
Proportion of reads containing the variant call (expect a value of approximately 0.5 for heterozygous calls and approximately 1 for homozygous calls) |
GT Depth |
Number of reads used for evaluating the corresponding call |
KNOWN Gene diseases |
Diseases known to be associated with variants in the gene, as annotated by ClinVar, HGMD, and OMIM |
Known pmid |
Pubmed ID of the reference from which the information was obtained |
KNOWN var diseases |
Diseases known to be associated with the variant as annotated by ClinVar, HGMD, and OMIM |
OMIM IDs |
Online Mendelian Inheritance in Man (OMIM) IDs |
rsIDs |
Reference SNP identifiers assigned by NCBI to a group of SNPs that map to an identical location |
Tip
Clicking the Info icon next to each column header opens a tooltip with the column description.
List view¶
Select tabs (e.g., Variants and Missing) can also be viewed in List view, which provides an abbreviated summary of the variant type along with key annotations. Annotations are numbered for reference in the figure below, followed by a table containing corresponding descriptions.
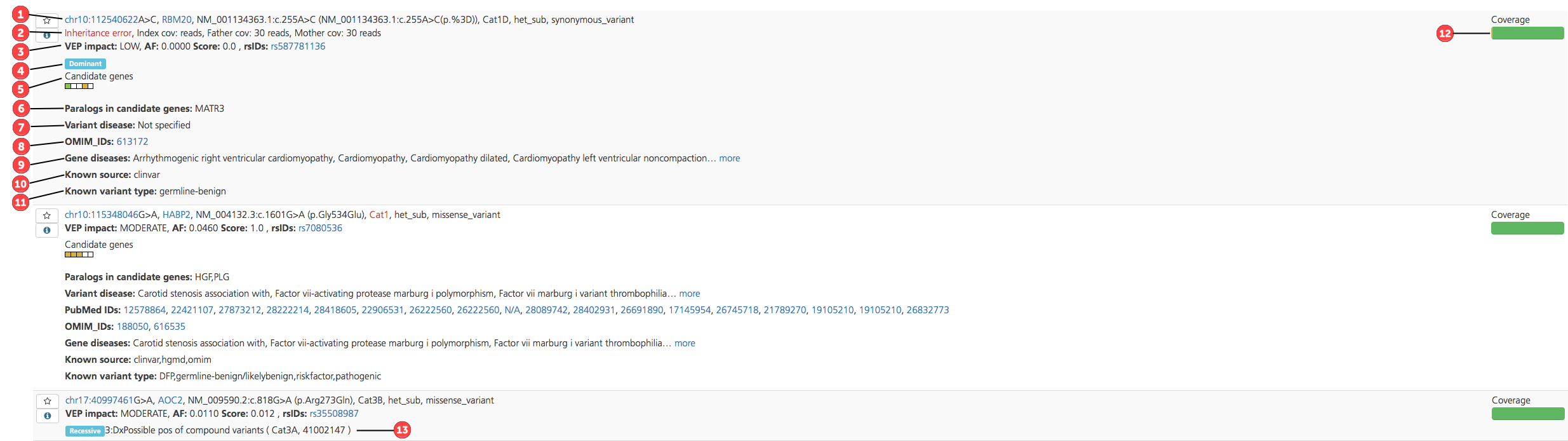
Figure reference |
Annotation |
Description |
|---|---|---|
1 |
Sequence Variant |
Variant location, reference variant, called variant, gene, coding sequence, protein position, and protein effect information (see Sequence Variant below) |
2 |
Inheritance Error |
If the identified variant does not appear to be transmitted in a Mendelian fashion based on the parental genotypes, the variant is flagged as an Inheritance error. Following the error message, the coverage (cov) is reported for the index, father, and mother to describe the read coverage in each participant. An inheritance error message can be caused by a number of factors, for example:
|
3 |
VEP impact (VEP max impact) |
Classification of the variant based on the severity of the consequence type on the transcript (“HIGH”, “MODERATE”, “LOW”, “LOWEST”); for a given variant, the maximum observed impact across the predicted transcripts from this position is reported (see also Variant Effect Predictor (VEP) Settings) |
AF (VEP max AF) |
Maximum reported population allele frequency derived from the 1000 Genomes Project phase 3 (1000G3), Exome Variant Server (EVS), Exome Aggregation Consortium (ExAC), Genomes of the Netherlands (GONL), and Kyoto population surveys (see also popAlleleFreq) |
|
Score (VEP max score) |
Each missense variant is assigned a score for the predicted impact of the amino acid change on protein function: maximum score for the variant as observed in dbNSFP (Score=max of (1-Sift_score), Polyphen2_HDIV_score, or Polyphen2_HVAR_score); scores run from 0 - 1; for other variant types, the VEP score is either displayed as 0.0 or omitted |
|
rsIDs |
Reference SNP identifiers assigned by NCBI to a group of SNPs that map to an identical location |
|
4 |
Inheritance |
Flags that indicate which inheritance model(s) the variant fits: Dominant, Recessive, or De Novo |
5 |
Candidate genes |
The Candidate genes ideogram displays color-coded boxes that correspond to the user-defined phenotypes, and that highlight which phenotypes or genelists match the candidate gene or a paralog of the candidate gene. Clicking the ideogram opens a pop-up window that lists the phenotypes in the order in which they appear in the ideogram. Color-codes are as follows:
|
6 |
Paralogs in candidate genes |
Known paralogs of the variant-containing gene also found in the candidate gene list; if the variant is in a gene that is a paralog of one or more Candidate genes, the candidate gene(s) are listed. |
7 |
Variant disease |
If the variant is known to be pathogenic, this field lists the diseases known to be caused by the variant; annotations are derived from HGMD, ClinVar, and OMIM databases |
8 |
OMIM and/or PubMed IDs |
OMIM and/or PubMed IDs associated with the variant |
9 |
Gene diseases |
If the variant gene is known to be pathogenic, this field lists the diseases known to be caused by the gene; annotations are derived from HGMD, ClinVar, and OMIM databases |
10 |
Known source |
|
11 |
Known variant type |
|
12 |
Coverage |
The gene coverage ideogram provides a graphical representation of the degree of read coverage across the gene that contains the variant. Coverage is represented as follows:
For emphasis, orange and red segments are displayed at double their proportionate size. In cases where <50% of the gene is covered by 10 or more reads, no green is displayed. The coverage calculation can be based on either the coding sequence or the entire gene including the 5’ and 3’ UTR, and can be based on either Ensembl or Refseq transcripts (these options are selected the Advanced report settings tab of the Study Overview). |
13 |
recessiveCat |
recessiveCat is a system for assessing recessive and compound heterozygous variants, based on the ACMG category for the two alleles (see recessiveCat below). |
Note
If no annotation is available for a particular field, the field is omitted from the Variant listing.
Sequence Variant and recessiveCat annotations are described in more detail below.
Sequence Variant¶
The Sequence Variant annotation provides positional, genotype, and protein effect information about the selected variant. Listed first is the variant’s genomic position based on the current reference assembly, followed by the gene symbol for the gene containing the variant.
Next, a description of the cDNA and protein position of the variant is given using a notation similar to the HGVS notation. In instances where a variant may affect one or more annotated transcripts and occur in different positions in the transcript, the position is given in parentheses followed by the DNA or protein change (e.g., “”c.(145,850)_C>T p.(284,49)_E>K,”). The DNA or protein change is given as “reference>sample”, and the altered nucleic acid sequence of the variant will follow the reference sequence. If the variant affects codons within a transcript, the position of the protein amino acid is also provided for the corresponding transcript listed. cDNA and HGVS notations are not used in the case of a splice variant.
Following the cDNA and protein position are the ACMG variant categorization (e.g., Cat1, Cat2), the zygosity and type of variant (e.g. het_sub, hom_ins, het_del), and the maximal VEP consequence (e.g., missense, splice_donor_variant).
Field header |
Description |
|---|---|
Chrom |
Variant chromosome location |
Pos |
Variant start bp position |
GENE Symbol |
HGNC gene symbol for gene identified (clone name provided when HGNC unavailable) |
CDS position |
Position of the base pair in the coding sequence; a value is given for each transcipt |
Ref |
Reference allele in VCF format at the designated position |
Call |
Sequence (variant) called based on the reference sequence at the designated position |
Protein Position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcipt specified in the CDS position field |
ACMGcat |
Categorization of the sequence variants according to the ACMG scheme |
Nucleotide Change |
Description of the nucleotide change in the Human Genome Variation Society (HGVS) nomenclature (e.g., deletion (del), substitution (sub), insertion (ins)) |
Consequence |
Impact of the variation on the sequence as calculated per transcript by NCBI (e.g., frameshift variant, truncating variant, missense variant, splice site variant) |
recessiveCat¶
To assess multiple variants in a single gene, Genuity Science has adapted the approach taken by Kingsmore, et al., 20111.
When a gene has two or more variants, each variant is given a category classification using the combined analysis of the variants in the gene.
The recessive and compound heterozygous category is based on the ACMG category for two alleles from any two variants observed in a gene or from a homozygous variant. For an allele combination in the index to be considered a potential recessive or compound heterozygous candidate, it may not be present in any of the controls (unless ctrlDelta is greater than zero). If the alleles in the index can be phased, they are considered only if they are not known to be from the same parent. In controls, the phase of alleles is assumed to be different.
Allele 1/Allele 2 |
Cat1 |
Cat2 |
Cat3a |
Cat3b |
Cat4 |
|---|---|---|---|---|---|
Cat1 |
1 |
1 |
2 |
3 |
5 |
Cat2 |
1 |
1 |
2 |
3 |
5 |
Cat3a |
2 |
2 |
3 |
3 |
6 |
Cat3b |
3 |
3 |
3 |
4 |
6 |
Cat4 |
5 |
5 |
6 |
6 |
7 |
“Consistent”, “Likely”, and “Unlikely” classifications are derived from the ACMG guidelines for variant categorization. In cases where, for example, one variant is known to be pathogenic (Cat1), and a variant in the second allele is a truncating variant (Cat2), the combined variants are considered “Consistent” with diagnosis. For a Cat2 variant and a Cat3a variant, the combined variant genotype would be classified as “DxLikely” to cause the phenotype. Two Cat1 variants would score the highest with a “DxConsistent” classification.
References
Kingsmore SF, Dinwiddie DL, Miller NA, Soden SE, Saunders CJ, The Children’s Mercy Genomic Medicine Team. Adopting orphans: comprehensive genetic testing of Mendelian diseases of childhood by next-generation sequencing. Expert review of molecular diagnostics. 2011;11(8):855-868. doi:10.1586/erm.11.70.
Starred variants¶
“Starring” a variant makes it easily retrievable for later review without the need for a comment.
To star a variant, click the Star icon in the leftmost column of a selected row in the Variant listing.

When a variant is starred, the star icon turns orange. To “unstar” the variant, click the Star icon again. Starred variants are also indicated in the Starred column of the Variant listing (see Advanced Report Columns).
Approved variants in other studies¶
If a variant has been annotated with the Variant review status “Approved and current” in another study in the project, a green circle appears below the Info icon in the leftmost column of the Variant listing.
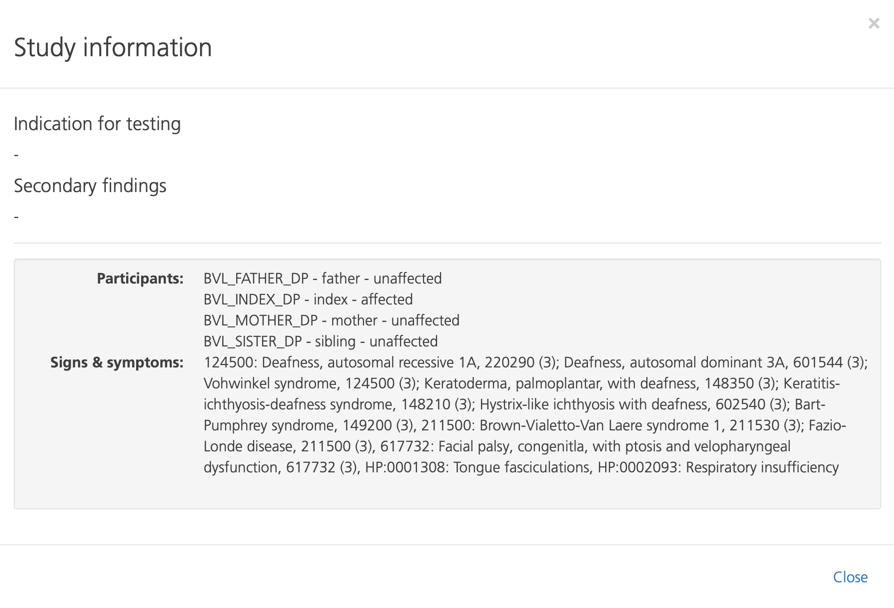
Viewing study information¶
Clicking Info at the top of the page opens the Study information pop-up window.
Saving the Variant listing¶
You can save the Variant listing by doing any of the following:
Click Open query in Sequence Miner at the top of the page. This loads the same set of variants and annotations in Sequence Miner. From Sequence Miner, you can cut and paste table rows into an application on your computer (limited to 10,000 rows).
Save the Variant listing to the user_data directory in the Sequence Miner File Explorer, and then copy and paste selected rows into an application on your computer.
Generate a tab-delimited file by adding .gor to the URL of the CSA page.