Editing the Variant Summary¶
The Variant Summary window is opened by clicking the Information icon next to a selected variant in the variant listing.
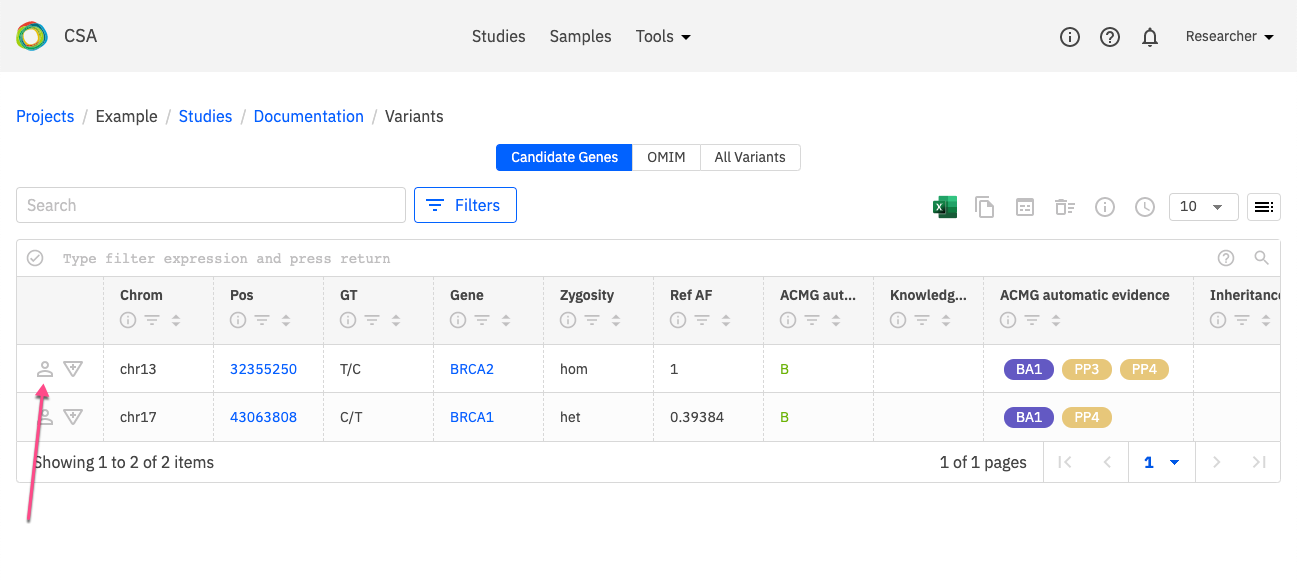
The Variant Summary consists of the following panels:
Variant summary header
Variant information
Variant quality
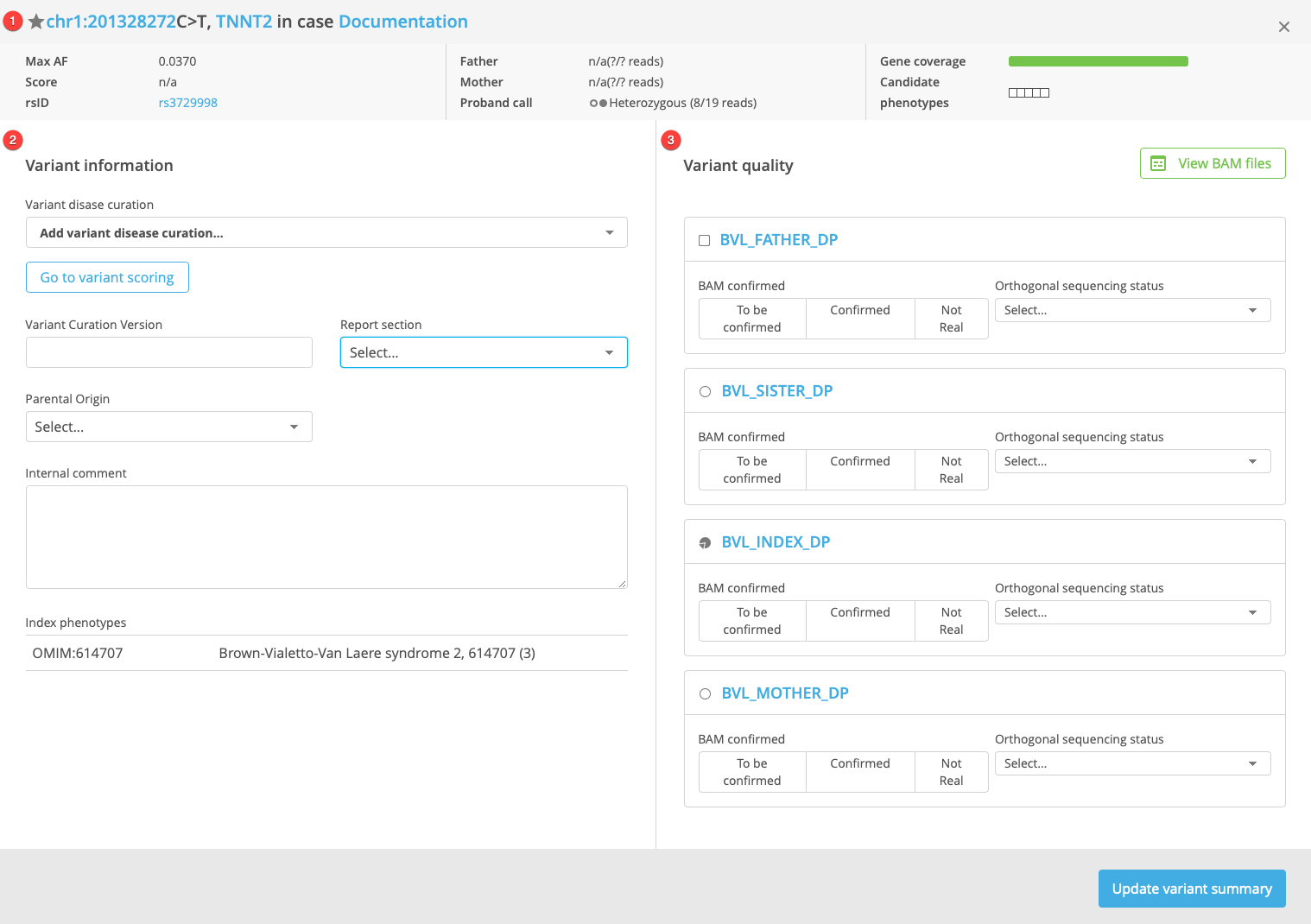
Each panel is described in more detail below.
Variant summary header¶
The Variant summary header displays various details of the variant in the context of the current case. These details include the variant identity, Max AF, Max score (derived from computational and predictive scores from the dbNSFP resource), rsID (dbSNP accession number), variant information for the proband and parents if available (including number of variant calls and total read depth from the VCF), gene coverage (average coverage across the current gene), and candidate phenotypes.
Variant information¶
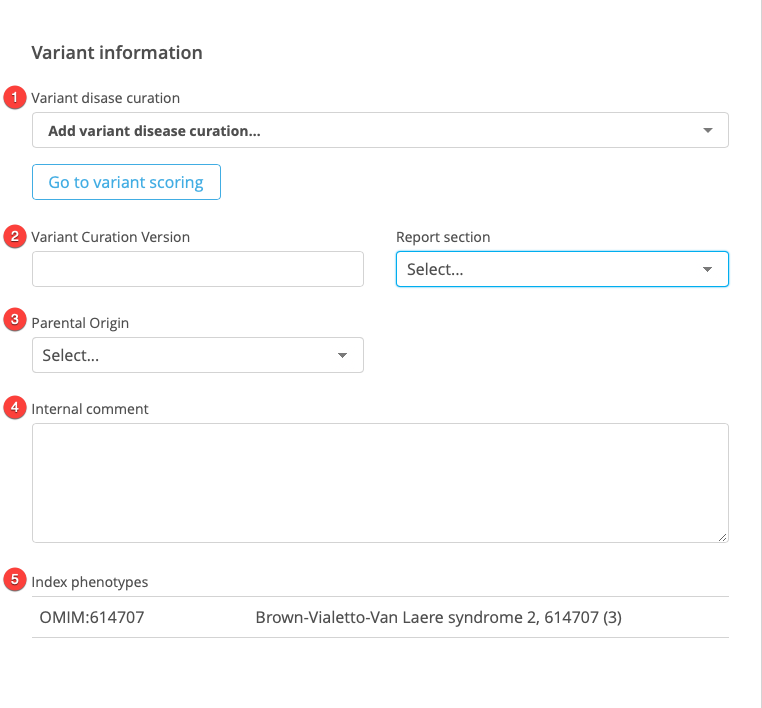
This Variant information panel includes the following fields:
Variant disease curation
Parental origin
Genomic origin
Report section
Internal sample comment
Index phenotypes
Variant disease curation¶
Open the drop-down menu to select from existing variant curations for the selected variant or to create a new variant curation.
Each variant curation is uniquely defined by VDT:
Variant identity (position, ref string, alternate string, gene, and genome build)
Disease
Transcript
Selecting New variant disease curation opens a New curation window, where disease and transcript can be selected and added to the curation. If the selected combination of VDT already exists, a prompt is displayed to redirect to the existing curation (see also Adding a new variant curation).
Selecting Clone next to an existing variant disease curation copies the annotations from the existing variant curation to a new variant curation.
Parental origin¶
Open the drop-down menu to select the Parental origin: “Father”, “Mother”, “Both”, “De novo”, or “Unconfirmed”. The value will be stored as part of the sample curation entry.
Genomic origin¶
Open the drop-down menu to select the Genomic origin: “Autosomal”, “x-linked”, “y-linked”, or “Mitochondrial”. The value will be stored as part of the sample curation entry.
Report section¶
Select whether the variant curation should be included in the clinical report and in which section the variant should appear. Options include “Not specified” (will not appear in the clinical report), “Primary”, “Secondary”, “Incidental”, or “Excluded” (will not appear in the clinical report).
Internal sample comment¶
Enter internal notes about the variant or case in the text box. Text entered here is saved as part of the sample curation entry and does not appear in the clinical report.
Note
It is not possible to add an internal sample comment without first updating the variant curation.
Index phenotypes¶
This section lists all of the phenotypes (or diseases - any HPO or OMIM terms) that are registered to the index case. Note that the phenotype and disease associations registered to the index case may be different from those registered to the study.
Variant quality¶
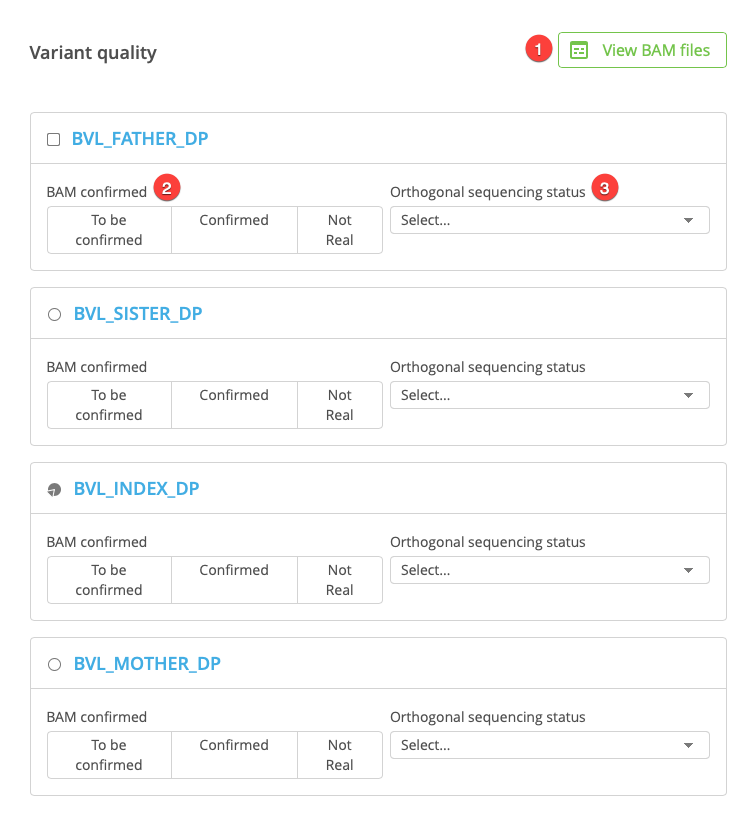
The Variant quality panel includes the following fields:
View BAM files
BAM confirmation
Orthogonal sequencing status
View BAM files¶
To view the aligned reads for the case in the Sequence Miner Genome Browser, click View BAM files at the top of the Variant quality panel. The Genome Browser automatically loads with all participants in the current study, and shows the current variant position.
BAM confirmation¶
Click the buttons to indicate that the BAM files have been reviewed and confirmed as Real or Not real. These values are saved as part of the sample curation for the corresponding individual.
Orthogonal sequencing status¶
Open the drop-down menu to select from the following options: “Not necessary”, “Confirmed real”, “Confirmed not real”, or “Results pending”. Selecting Results pending adds the variant to the Orthogonal sequencing confirmation tab on the Study Overview page.